Start treatment with CRYSVITA® as early as possible after diagnosis of XLH in suitable patients
Table of Contents
As of Sep 2020, CRYSVITA® has been approved by Korea MFDS (Ministry of Food and Drug Safety) for the treatment of FGF-23 related hypophosphatemic rickets and osteomalacia.1.
To decide if this is a suitable option for any of your patients, please click here for more information.
Product is approved in EU/US/Japan and selected markets in Asia Pacific. Local approved prescribing information may differ. Please refer to local approval status and prescribing information before prescribing.
Dosing
CRYSVITA® dosing is every 2 weeks in paediatric patients in FGF23 related hypophosphatemic rickets (except for tumor-induced osteomalacia).1
As of 3 May 2021, CRYSVITA has been approved by Health Science Authority (HSA) Singapore. To decide if this is a suitable option for any of your patients, please click here for more information.
Product is approved in EU/US/Japan and selected markets in Asia Pacific. Local approved prescribing information may differ.
Please refer to local approval status and prescribing information.
Children: This drug is subcutaneously administered at 0.8 mg/kg once for 2 weeks.
The dose may be increased or decreased as needed according to serum phosphorus concentration and symptoms, and the maximum dose should be 2 mg/kg per dose. However, one dose should not exceed 90 mg.
The starting dose and dose adjustment of this drug should be determined with reference to the following table. (Please see the table in the (‘Crysvita Prescribing Information‘.) The dose of this drug can be determined by rounding off the value calculated by body weight in the unit of 10 mg.
At the start of administration of this drug and dose adjustment, serum phosphorus concentration should be measured at every dosing time (once per 2 or 4 weeks) until serum phosphorus concentration becomes stable.
When this drug is to be administered to patients with FGF23-related hypophosphatemic rickets, if the bone age reaches 17 years in male and 15 years in female, the same dosage and administration as those for adults should be used according to the condition of patient.
If an oral phosphorus formulation or activated vitamin D3 formulation is administered, these drugs should be discontinued one week before starting this drug, and administration of this drug should be initiated after confirming the serum phosphorus concentration falls below the reference lower limit.
CRYSVITA® dosing is every 4 weeks in adult patients in FGF23 related hypophosphatemic rickets and Tumor-Induced Osteomalacia.1
1. FGF23-related hypophosphatemic rickets and osteomalacia (except for tumor-induced osteomalacia)
One dose should not exceed 90 mg. The dose may be decreased as needed according to serum phosphorus concentration and symptoms.
The starting dose and dose adjustment of this drug should be determined with reference to the following table. (Please see the table in the (‘Crysvita Prescribing Information‘.) The dose of this drug can be determined by rounding off the value calculated by body weight in the unit of 10 mg.
At the start of administration of this drug and dose adjustment, serum phosphorus concentration should be measured at every dosing time (once per 2 or 4 weeks) until serum phosphorus concentration becomes stable.
When this drug is to be administered to patients with FGF23-related hypophosphatemic rickets, if the bone age reaches 17 years in male and 15 years in female, the same dosage and administration as those for adults should be used according to the condition of patient.
If an oral phosphorus formulation or activated vitamin D3 formulation is administered, these drugs should be discontinued one week before starting this drug, and administration of this drug should be initiated after confirming the serum phosphorus concentration falls below the reference lower limit.
2. Tumor-induced osteomalacia
The dose may be increased or decreased as needed according to serum phosphorus concentration and symptoms, and the maximum dose should be 2 mg/kg per dose.
The starting dose and dose adjustment of this drug should be determined with reference to the following table. (Please see the table in the ‘Crysvita Prescribing Information‘.) The dose of this drug can be determined by rounding off the value calculated by body weight in the unit of 10 mg.
At the start of administration of this drug and dose adjustment, serum phosphorus concentration should be measured at every dosing time (once per 2 or 4 weeks) until serum phosphorus concentration becomes stable.
If an oral phosphorus formulation or activated vitamin D3 formulation is administered, these drugs should be discontinued one week before starting this drug, and administration of this drug should be initiated after confirming the serum phosphorus concentration falls below the reference lower limit.
Calculation for starting dose
Sample Starting Dose Calculation – Pediatric
Patient weight (kg) x Recommended starting dose (0.8mg/kg)
Example: 23kg x 0.8mg/kg = 18.4 mg (Round to nearest 10 mg)
Starting dose CRYSVITA® = 20 mg
Sample Starting Dose Calculation – Adult patient (for FGF23 related hypophosphatemic rickets)
Patient weight (kg) x Recommended starting dose (1mg/kg)
Example: 77kg x 1mg/kg = 77 mg (Round to nearest 10 mg)
Starting dose CRYSVITA® = 80 mg
Sample Starting Dose Calculation – Adult patient (for Tumor-Induced Osteomalacia)
Patient weight (kg) x Recommended starting dose (0.3mg/kg)
Example: 72kg x 0.3mg/kg = 21.6 mg (Round to nearest 10 mg)
Starting dose CRYSVITA® = 20 mg
Pediatrics Dose Adjustments1
Dose Increase for paediatric patients:
Normally administration is initiated in children according to the weight of patients by referring to the following table. Thereafter, the dose should be increased or decreased as needed according to serum phosphorus concentration and symptoms.
If the dose needs to be increased, for example, when the serum phosphorus concentration falls below the reference lower limit, the dose may be increased stepwise within the dose range up to 2 mg/kg or 90 mg per dose, whichever is lower. However, the dose increase should be at intervals of 4 weeks or longer.
At the start of administration of this drug and dose adjustment, serum phosphorus concentration should be measured at every dosing time (once per 2 or 4 weeks) until serum phosphorus concentration becomes stable.
Starting doses for pediatric patients with FGF23-related hypophosphatemic rickets and osteomalacia (except for tumor-induced osteomalacia)
| Body Weight (kg) | Starting Dose (mg) |
|---|---|
| 7-18 | 10 |
| 19-31 | 20 |
| 32-43 | 30 |
| 44-56 | 40 |
| 57-68 | 50 |
| 69-81 | 60 |
| 82-93 | 70 |
| 94-106 | 80 |
| 107 and greater | 90 |
Dose decrease (pediatric patients 1 to less than 18 years of age)1
Dose Decrease for paediatric patients:
Normally administration is initiated in children according to the weight of patients by referring to the above table. Thereafter, the dose should be increased or decreased as needed according to serum phosphorus concentration and symptoms.
If serum phosphorus concentration increases over the reference upper limit, the drug should be withdrawn until the serum phosphorus concentration falls below the reference lower limit. For the resumption of administration, the dose should be decreased roughly at half of the dose before withdrawal, and administration be resumed.
Adult Dose Adjustments1
Dose Decrease for adult patients with FGF23-related hypophosphatemic rickets and osteomalacia (except for tumor-induced osteomalacia)
Normally administration is initiated in adults according to the weight of patients by referring to the following table. Thereafter, the dose should be decreased as needed according to serum phosphorus concentration and symptoms.
If serum phosphorus concentration increases over the reference upper limit, the drug should be withdrawn until the serum phosphorus concentration falls below the reference lower limit. For the resumption of administration, the dose should be decreased roughly at half of the dose before withdrawal, and administration be resumed.
Starting doses for adult patients with FGF23-related hypophosphatemic rickets and osteomalacia (except for tumor-induced osteomalacia)
| Weight | Starting dose |
|---|---|
| 35 - 44 kg | 40 mg |
| 45 - 54 kg | 50 mg |
| 55 - 64 kg | 60 mg |
| 65 - 74 kg | 70 mg |
| 75 - 84 kg | 80 mg |
| 85 kg and above | 90 mg |
Dose Increase & Decrease for adult patients with Tumor-Induced Osteomalacia
Normally administration is initiated in adults according to the weight of patients by referring to the following table. Thereafter, the dose should be increased or decreased as needed according to serum phosphorus concentration and symptoms. If the dose needs to be increased, for example, when the serum phosphorus concentration falls below the reference lower limit, the dose may be increased stepwise within the dose range up to 2 mg/kg per dose.
At the start of administration of this drug and dose adjustment, serum phosphorus concentration should be measured at every dosing time (once per 2 or 4 weeks) until serum phosphorus concentration becomes stable.
If serum phosphorus concentration increases over the reference upper limit, the drug should be withdrawn until the serum phosphorus concentration falls below the reference lower limit. For the resumption of administration, the dose should be decreased roughly at half of the dose before withdrawal, and administration be resumed.
Starting doses for patients with tumor-induced osteomalacia’
| Weight | Starting dose |
|---|---|
| 17 - 49 kg | 10 mg |
| 50 - 83 kg | 20 mg |
| 84 - 116 kg | 30 mg |
Administration
CRYSVITA® Administration1
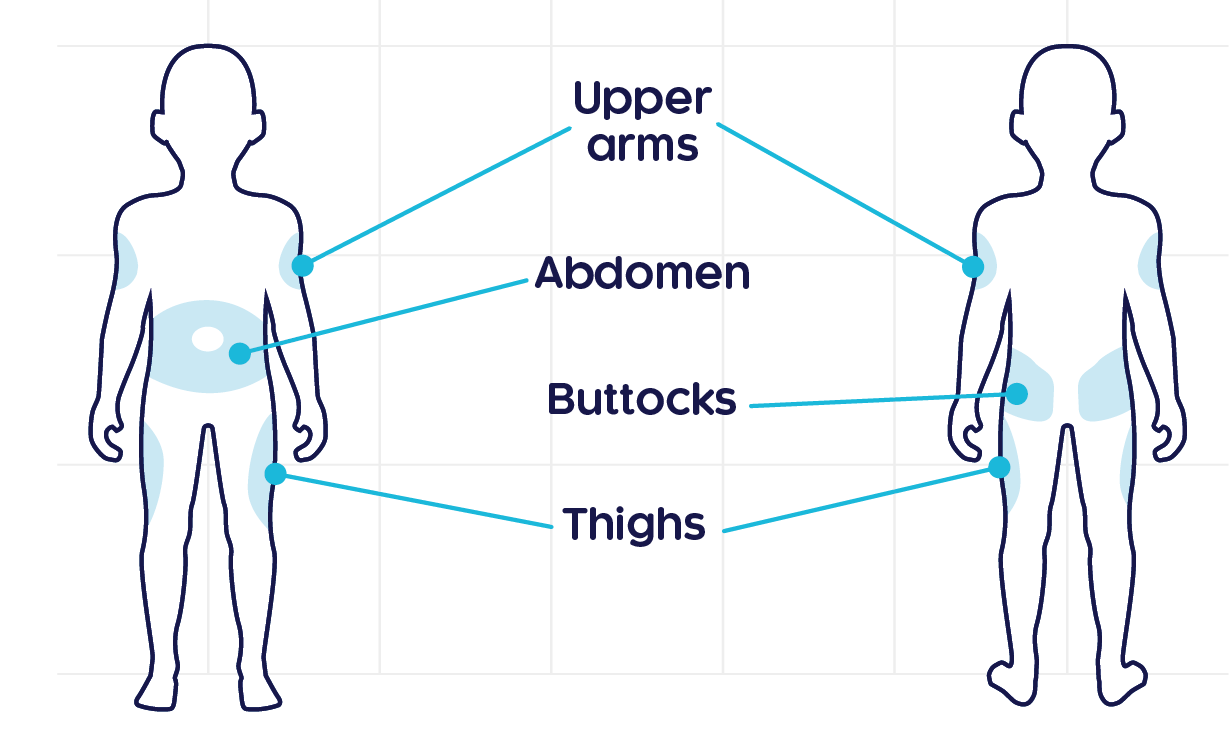
CRYSVITA® should be administered subcutaneously1
If an oral phosphorus formulation or activated vitamin D3 formulation is administered, these drugs should be discontinued one week before starting this drug, and administration of this drug should be initiated after confirming the serum phosphorus concentration falls below the reference lower limit.
Precautions in administration of drugs
- The maximum liquid amount to be administered per injection site should be 1.5 mL.
- This drug is for single use, and the residual liquid after use should not be used.
Administration of this drug should be initiated by physician in medical institution, or under the direct supervision of physician. The following precautions should be observed when this drug is to be taken by self-administration:
- Physicians should deliberately investigate the validity (no need of immediate dose modifications etc) of applying self-administration, provide patients or their guardians with necessary education and training, and confirm that patients or their guardians can surely administer the drug before allowing self-administration under their management and guidance.
- Careful monitoring including measuring serum phosphate levels should be continued for patients under self-administration condition.
- If adverse drug reactions due to this drug are suspected after application or it may be difficult to continue self-administration, self-administration should be immediately discontinued, and appropriate measures such as deliberate monitoring under management of physician should be taken.
- Patients and their guardians should be explained about adverse drug reactions and how to deal with them. They should be advised to contact their medical institutions if occurrence of any adverse drug reaction is suspected.
- Patients and their guardians should be alerted not to reuse the used syringe, and thoroughly instructed about safe disposal method.
Monitoring
Recommended monitoring schedule for CRYSVITA® patients2
| Assessment | Frequency |
|---|---|
| Fasting serum phosphate† | Month 1: Every 2 weeks |
| Month 2-3: Every 4 weeks | |
| Thereafter: As appropriate | |
| Following dose adjustment: 4 weeks after | |
| Renal ultrasonography | At least every 2 years in patients without nephrocalcinosis and at yearly intervals in patients with nephrocalcinosis and/or persistent hypercalciuria |
| Plasma ALP, calcium, PTH and creatinine | In children aged 1–5 years: Every 1-3 months, or as indicated |
| In children 5-12 years: Every 3-6 months, or as indicated | |
| Urine calcium and phosphate‡ | Every 3 to 6 months |
†It is recommended that fasting serum phosphate is targeted at the lower end of the normal reference for age.
‡Upper normal range (mol/mol): 2.2 (<1 years), 1.4 (1–3 years), 1.1 (3–5 years), 0.8 (5–7 years) and 0.7 (>7 years).
Contraindications, warnings and precautions1
Contraindications
Do not administer to the following patients
- Patients on medication of oral phosphorus formulation or activated vitamin D3 formulation
- Patients with severe renal impairment or end stage renal failure. The risk may be particularly high for developing hyperphosphataemia and calcification in organs such as the kidney. No clinical studies have been conducted in this patient population
- Patients with previous hypersensitivity to the ingredient of this drug
- For FGF23-related hypophosphatemic rickets and osteomalacia associated with the administration of saccharated iron oxide and polymaltose iron, this drug must not be administered, and consideration should be made to discontinue treatment with any drugs that may cause excessive FGF23 production.
Warnings
The following patients should be administered with care
- Patients with complication or history of diseases, etc.
- Patients with hypercalcaemia : Hypercalcaemia may worsen.
- Patients with moderate or mild renal impairment
During administration of this drug, patients should be evaluated for the renal function on a regular basis to be qualified for the treatment. In addition, attention should be paid to changes in the serum phosphorus concentration.
Special Populations (Pregnancy, Delivery or Lactation)
- This drug should be administered to pregnant women or women suspected of being pregnant, only if the therapeutic benefits are thought to outweigh any possible risks. Results of a reproductive and developmental toxicity study in monkeys demonstrated an increase in preterm birth rate at a dose corresponding to 3.7-fold of the exposure at the clinical maximum dose, an increase in the placental weight and mineral deposition, and an increase in the abortion rate and embryonic/fetal mortality at a dose corresponding to 32-fold of the exposure at the clinical maximum dose.
- Continuation or discontinuation of breast feeding should be discussed considering therapeutic benefits and benefits of breastfeeding. Transfer of this drug to milk is unknown.
Storage
CRYSVITA® Storage1
Precautions in preparation of drugs
- Drugs should be taken out from the refrigerator to make into room temperature before administration.
- An appropriate small syringe should be selected for accurate suction of liquid amount necessary for administration.
- Do not mix with other drugs for injection.
Precautions in Storage
- Keep the vial in the original carton to protect from light until time of use.
- Do not freeze or shake.
- Please keep out of reach of children
- Light-resistant storage after opening outer box
- Do not put it in another container to prevent confusion in storage
Storage
- Stored at 2-8°C in light-resistant condition, Hermetic container
- Do not use after the expiration date indicated on the package
Please refer to the full Prescribing Information for full details before prescribing.
1. CRYSVITA® (burosumab). Based on Korean Insert paper. Kyowa Kirin Asia Pacific Pte Ltd; May 2021 2. Haffner D et al. Nat Rev Nephrol. 2019; 15(7): 435–455.