XLH는 드물게 발생하는 유전성*, 진행성, 일생 동안 지속되는 인산 소실
질환이며, FGF23 과잉을 초래하는 PHEX 유전자에 생긴 돌연변이에
의해 발생됩니다.1-4
Table of Contents
XLH의 원인
XLH는 FGF23 레벨 증가로 인한 만성 저인산혈증이 나타나는 것이 특징입니다2,4
인산염은 여러 생물학적 기능에서 중요합니다.5 정상적인 인산염 항상성이 손상되면 중증의 합병증이 나타나며 전반적인 건강에 유해한 영향을 미칩니다.5
XLH에서, FGF23 레벨이 과잉 상태이면 신장에서 인산염 재흡수와 활성 비타민D 1,25(OH)2D 생성이 감소하고, 소변을 통한 인산염 배설이 증가하며 장내 인산염 흡수가 감소합니다. 이로 인해 혈청 인산염 레벨이 저하됩니다.2.6

과잉 FGF23 레벨은 만성 저인산혈증을 유발하여, 골 무기질화가 손상되며, 그로 인해 XLH가 있는 소아의 성장과 발달에 심각한 영향을 미치며 XLH가 있는 성인에서 골연화증이 초래됩니다.8
임상 증상
XLH는 뼈, 근육 및 치아에 영향을 미치는 다기관 질환입니다.4
XLH의 임상적 발현증상은 일생 중 매우 초기에 나타나며, 전형적으로 하지 변형(예: 다리 휨)이 처음 2년 동안 명백해집니다.3,4
소아 XLH 환자는 구루병, 진행성 하지 변형, 성장 손상, 건강하지 못한 치아 상태를 나타내며, 어떤 경우에는 두개골 변형을 유발하지만, 환자마다 징후와 증상의 중증도가 다를 수 있습니다.6
성인 XLH 환자의 증상은 뼈, 관절 및 연골 결손으로 인한 골절/가성골절, 조기 발생 골관절염, 골극(osteophyte) 형성, 부착부 병증(enthesopathies) 및 척추 협착증이 있습니다. 성인은 치아 손상, 감각 신경성 난청, 이명 및 현기증을 경험할 수 있습니다.6
XLH 환자는 흔히 운동성(mobility)과 신체 기능 손상, 뼈와 관절 통증, 피로 및 일생 동안 정서적 및 사회적 손상을 나타냅니다.4,8
XLH의 임상적 증상은 심각도가 다르고 다양하게 발현되며, 신체적 기능 및 삶의 질에 영향을 줄 수 있습니다.6,8,9
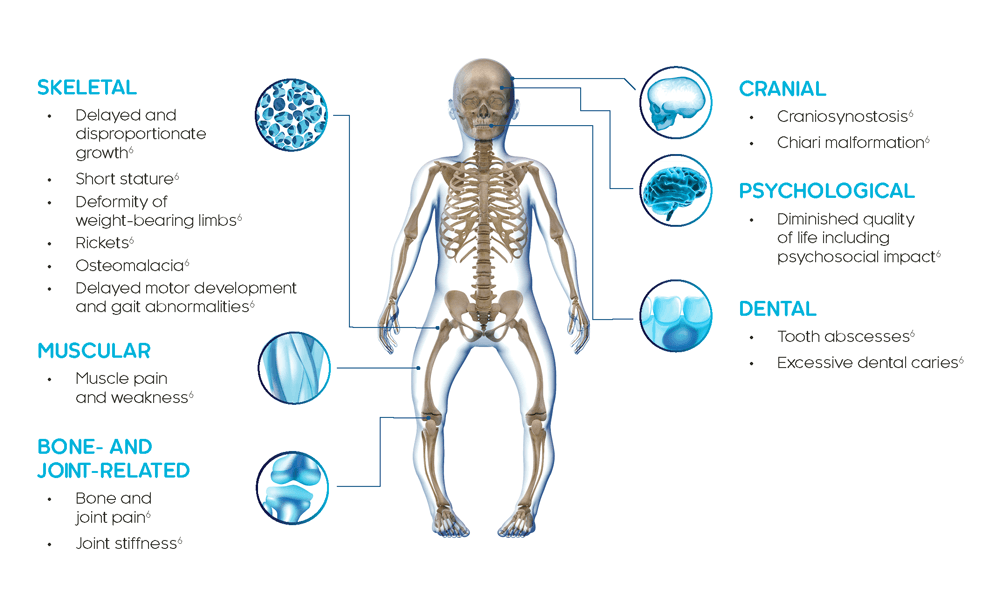
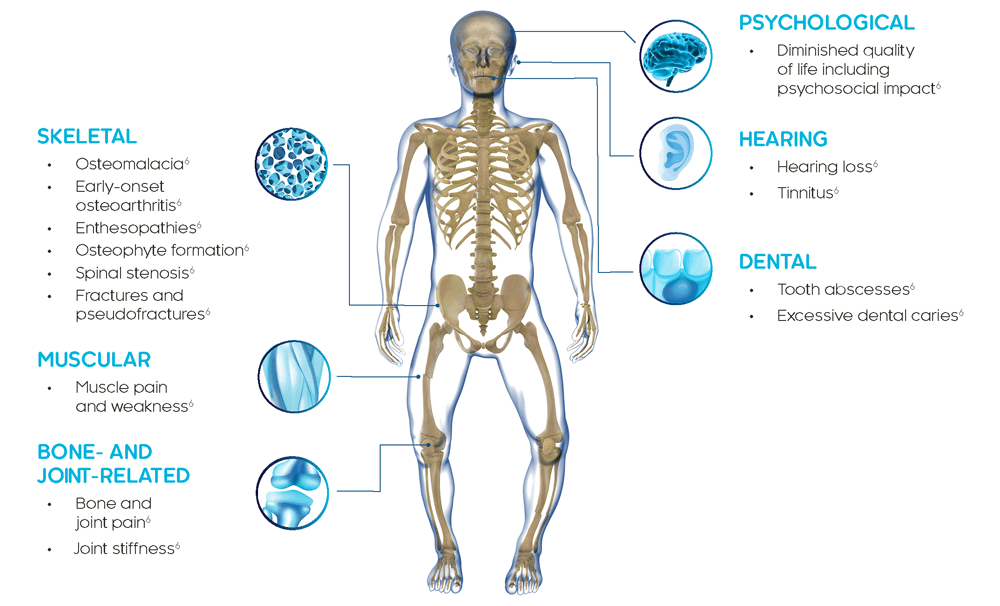
XLH는 일생 동안 지속되는 진행성 질환이며 임상적 발현증상은 시간이 경과됨에 따라 축적됩니다. XLH가 있는 성인은 소아기부터 이어져온 임상적 발현증상과 함께 나중에 나타나는 새로운 합병증을 나타냅니다.8
유병률
XLH는 드물게 발생하는 희귀질환입니다.
대한민국에서 희귀질환이란 유병인구가 2만명 이하이거나 진단이 어려워 유병인구를 알 수 없는 질환으로 보건복지부령으로 정한 절차와 기준에 따라 정한 질환을 말합니다.10
XLH는 대략 인구 20,000명당 또는 60,000명당 1명 발생하는 것으로 추정됩니다. 1,6,7
질환의 희귀성(rarity) 때문에, XLH 진단과 치료가 종종 지연되는 경우가 발생합니다.4
유전 형식
XLH는 X 염색체 우성 형식으로 유전됩니다. 그러나, 환자의 20-30%는 자연발생 돌연변이로 인해 발생 됩니다11–13
XLH는 PHEX 유전자의 돌연변이로 인해 발생됩니다.4 대부분의 경우, XLH는 X 염색체 우성 형식으로 유전됩니다.11
XLH의 유전형식
영향을 받은 부모와 그로 인해 영향을 받는 아들과 딸의 비율을 확인하려면 아래에서 아버지 또는 어머니를 클릭하십시오.
(짙은 파란색은 영향을 받는 사람을 나타냅니다).


유전자 분석은 XLH의 진단을 확인하고 위험 발생 가능성이 있는 가족 구성원을 파악하는데 도움이 될 수 있습니다.4
*XLH의 20~30%는 가족력 없이 자연적 으로 발생할 수 있습니다.12,13
1. Beck-Nielsen SS, et al. Eur J Endocrinol. 2009;160:491–97. 2. Martin A & Quarles LD. Adv Exp Med Biol. 2012;728:65–83. 3. Carpenter TO, et al. J Bone Miner Res. 2011;26:1381–88. 4. Haffner D, et al. Nat Rev Nephrol. 2019;15:435–55. 5. Penido MGMG & Alon US. Pediatr Nephrol. 2012;27:2039–48. 6. Beck-Nielsen SS, et al. Orphanet J Rare Dis. 2019;14:58. 7. Rafaelsen S, et al. Eur J Endocrinol. 2016;174:125–36. 8. Skrinar A, et al. J Endocr Soc. 2019;3:1321–34. 9. Linglart A, et al. Endocr Connect. 2014;3:R13–30. 10. Rare Disease Fund to Provide Financial Support to Singaporeans with Rare Disease. Available at: https://www.moh.gov.sg/news-highlights/details/rare-disease-fund-to-provide-financial-support-to-singaporeans-with-rare-diseases Accessed: December 2021. 11. Whyte MP, et al. J Clin Endocrinol Metab. 1996;81:4075–80. 12. Rajah J, et al. Eur J Pediatr. 2011;170:1089–96. 13. Dixon PH, et al. J Clin Endocrinol Metab. 1998;83:3615–23.