XLH is a rare, hereditary,* progressive and life-long phosphate wasting disorder, caused by mutations in the PHEX gene that leads to excess FGF231-4
Table of Contents
Causes of XLH
XLH is characterised by chronic hypophosphataemia due to increased FGF23 levels2,4
Phosphate is critical to many biological functions.5 Disruption of normal phosphate homeostasis is associated with severe complications and has detrimental effects on overall health.5
In XLH, excessive levels of FGF23 decrease renal phosphate reabsorption and active vitamin D (1,25[OH]2D) production, which increases urinary phosphate excretion and reduces intestinal phosphate absorption, respectively; this leads to low serum phosphate levels.2,6

Excess FGF23 leads to chronic hypophosphataemia resulting in impaired bone mineralisation, severely affecting the growth and development of children with XLH2,6 and leading to osteomalacia in adults with XLH.8
Clinical manifestations
XLH is a multisystemic disease that impacts bone, muscle and dentition4
The clinical manifestations of XLH develop very early in life and usually become apparent during the first 2 years typically presenting as lower limb deformities (e.g. bowing of the legs).3,4
In children, XLH causes rickets, progressive lower limb deformity, impaired growth, poor dental health and, in some cases, skull deformities, but the signs and symptoms can vary in severity.6
In adults with XLH, bone, joint and cartilage deficits include pseudofractures/fractures, early-onset osteoarthritis, osteophyte formation, enthesopathies and spinal stenosis. Adults can experience dental impairments, sensorineural hearing loss, tinnitus and vertigo.6
People with XLH commonly exhibit impairments in mobility and physical function, bone and joint pain, fatigue as well as emotional and social detriments throughout life.4,8
The clinical manifestations of XLH vary in presentation and severity, and can impair physical function and quality of life6,8,9
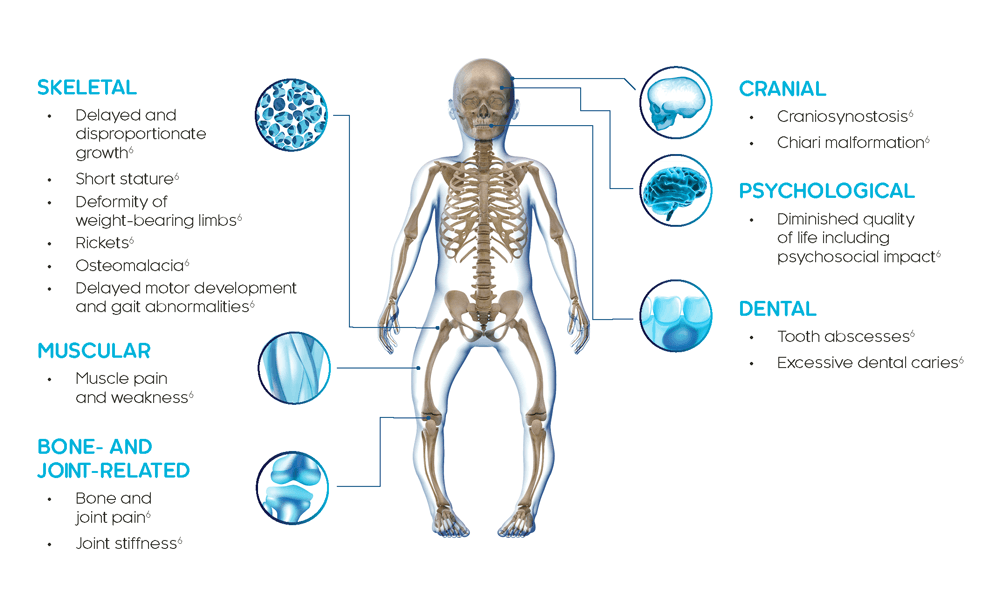
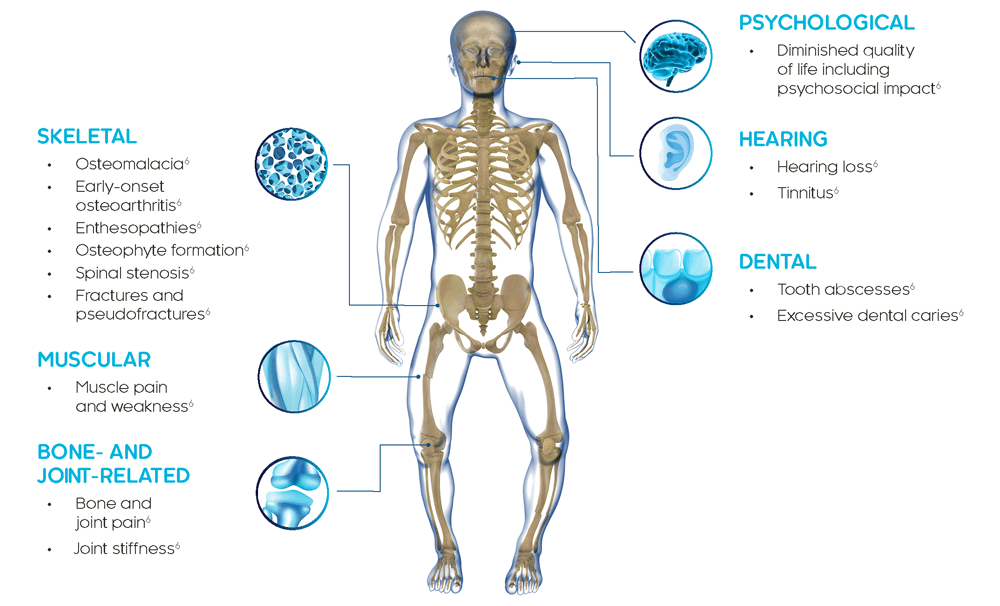
XLH is a progressive, life-long disease and clinical manifestations accumulate over time. Adults with XLH present with clinical manifestations that derive from childhood as well as new complications that occur later in life.8
Prevalence
XLH is a rare disease
In Singapore, the Ministry of Health of Singapore (“MOH”) defined a rare disease as a condition that affects fewer than 1 in 2,000 people.10
XLH is estimated to affect approximately 1 in 20,000– 1 in 60,000 people.1,6,7
Due to its rarity, there is often a delay in diagnosis and treatment.4
Hereditary pattern
XLH is inherited in an X-linked dominant pattern; however, 20–30% of cases arise from spontaneous mutations11–13
XLH is caused by mutations in the PHEX gene.4 In most cases, XLH is inherited in an X-linked dominant pattern.11
Hereditary pattern of XLH
Click the father or mother below to reveal the ratio of affected sons and daughters with an affected parent. (dark blue indicates affected individual).


Genetic analysis can help confirm a diagnosis of XLH and identify family members who may be at risk.4
*In approximately 20–30% of cases, XLH occurs spontaneously and there is no family history.12,13
1. Beck-Nielsen SS, et al. Eur J Endocrinol. 2009;160:491–97. 2. Martin A & Quarles LD. Adv Exp Med Biol. 2012;728:65–83. 3. Carpenter TO, et al. J Bone Miner Res. 2011;26:1381–88. 4. Haffner D, et al. Nat Rev Nephrol. 2019;15:435–55. 5. Penido MGMG & Alon US. Pediatr Nephrol. 2012;27:2039–48. 6. Beck-Nielsen SS, et al. Orphanet J Rare Dis. 2019;14:58. 7. Rafaelsen S, et al. Eur J Endocrinol. 2016;174:125–36. 8. Skrinar A, et al. J Endocr Soc. 2019;3:1321–34. 9. Linglart A, et al. Endocr Connect. 2014;3:R13–30. 10. Rare Disease Fund to Provide Financial Support to Singaporeans with Rare Disease. Available at: https://www.moh.gov.sg/news-highlights/details/rare-disease-fund-to-provide-financial-support-to-singaporeans-with-rare-diseases Accessed: December 2021. 11. Whyte MP, et al. J Clin Endocrinol Metab. 1996;81:4075–80. 12. Rajah J, et al. Eur J Pediatr. 2011;170:1089–96. 13. Dixon PH, et al. J Clin Endocrinol Metab. 1998;83:3615–23.